Actualización 1 (17/12/2018):
-Se explicó otro camino para graficar utilizando ggplot2
Actualización 2 (18/12/2018)
-Se añadió el valor del coeficiente de correlación (r) dentro del gráfico empleando ggplot2
En el día de hoy aprenderemos a hacer un test de correlación Mantel en R utilizando el paquete vegan.
-Se explicó otro camino para graficar utilizando ggplot2
Actualización 2 (18/12/2018)
-Se añadió el valor del coeficiente de correlación (r) dentro del gráfico empleando ggplot2
En el día de hoy aprenderemos a hacer un test de correlación Mantel en R utilizando el paquete vegan.
¿En qué consiste este test? Básicamente lo que hace es estimar si hay o no correlación ya sea positiva o negativa entre dos conjuntos de datos de distancias (matrices). Por ejemplo: podemos probar si hay o no correlación entre la distancia genética y geográfica entre poblaciones de alguna especie (de esta manera se evalúa si hay aislamiento genético por distancia); o también podemos evaluar si existe o no correlación entre la disimilitud de comunidades biológicas y la distancia geográfica entre ellas.
Lo primero que debemos hacer es conocer cómo vamos a organizar los datos para introducirlos a R. Supongamos que tenemos 5 poblaciones (A-E) de Aedes albopictus. Entonces en teoría deberíamos considerar 10 comparaciones teóricas (5*(5-1)/2). Es importante que al momento de construir nuestra matriz coloquemos el mismo orden de las poblaciones tanto en la primer columna como en la primera fila (y que el orden sea igual en ambas matrices). Como verán así obtendremos una matriz cuadrada; y por supuesto, una matriz simétrica; ya que, al transponer las distancias de la segunda columna, estos serán equivalentes a los valores de la segunda fila. Los valores de la diagonal serán 0 (puesto que, la comparación se realiza entre la misma población). Veamos:
A B C D E
A 0 0.9 0.7 0.3 0.8
B 0.9 0 Terminen de rellenar ustedes... así le agarran el tiro ;)
C 0.7 0.5 0
D 0.3 0.1 0.7 0
E 0.8 0.2 0.6 0.3 0
Una vez entendido esto.. empecemos...
Para este ejemplo utilizaré unos datos compartidos por mi profesor de Ecología II para una práctica de laboratorio. Les explico rápidamente de qué van estos datos. Se tratan del muestreo de invertebrados asociados a 11 boñigas (cada boñiga es considerada como una comunidad). La pregunta es la siguiente: ¿La similitud de composición de especies depende de la distancia entre las boñigas?
Para esto se construyeron dos matrices (descargar acá):
1. Matriz de disimilitud (disim.txt) empleando el complemento del índice de Chao-Jaccard.
2. Matriz de distancia geográfica (geo.txt) entre las boñigas
#Una vez tengamos las matrices descargadas, procedemos a abrir R e instalamos el paquete:
install.package("vegan")
#Lo cargamos
library(vegan)
#Verificamos nuestro directorio de trabajo
getwd()
"C:/Users/Admin/Documents"
#Copiamos las matrices y las pegamos en esa ruta
#Cargamos matrices:
geo <- read.table("geo.txt", header=T)
disimi <- read.table("disim.txt", header=T) #El argumento header nos permite especificarle a la función que la primera fila y columna de nuestro archivo contienen los nombres de las variables
#Aquí ustedes ya pueden hacer el test, simplemente hacen esto:
test <- mantel(geo, disimi, method= "pearson", permutations= 1000) #Ustedes pueden elegir otro método, ya sea Kendall o Spearman. También pueden cambiar el número de permutaciones
Mantel statistic based on Pearson's product-moment correlation
Call:
mantel(xdis = distgeo, ydis = disimilitud, method = "pearson", permutations = 1000)
Mantel statistic r: 0.3423
Significance: 0.031968
Upper quantiles of permutations (null model):
90% 95% 97.5% 99%
0.198 0.296 0.355 0.423
Permutation: free
Number of permutations: 1000
#Gráfico de dispersión#
#Primero le indicamos a R que nuestros datos cargados corresponden a matrices de distancia:
matrizgeo <- as.matrix(geo)
matrizsim <- as.matrix(disimi)
#Graficamos
grafica <- plot(matrizgeo, matrizsim, pch=16, cex=1, col="black", xlab="Distancia geográfica (m)", ylab="Disimilitud de composición de especies")
grafica
#Obtendremos algo así:

Otro camino para graficar con ggplot2
install.packages("ggplot2")
install.packages("extrafont")
#Cargamos los paquetes
library(ggplo2)
library(extrafonts)
#Creamos un data frame para extraer los valores de las matrices, ya sea la fracción triangular superior o inferior (en este ejemplo, utilizaremos la inferior, es decir: lower)
df <- data.frame(Disimilitud=disimi[lower.tri(disimi)], Distancia=geo[lower.tri(geo)])
#Revisamos df
df
#¿Ven lo que hicimos? hemos organizado las variables de manera pareada (55 comparaciones en total)
##Comenzamos a graficar##
plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point()

plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman")

#Modificamos los títulos de los ejes con la función labs
plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud")

plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud") + theme(axis.title = element_text(face="bold"))

plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud") + theme(axis.title = element_text(face="bold"), axis.text = element_text(colour="black"))

#Podemos añadir la línea de tendencia con la función geom_smooth
plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud") + theme(axis.title = element_text(face="bold"), axis.text = element_text(colour="black")) + geom_smooth(method=lm)

#Si quieren remover el intervalo de confianza, modificamos el argumento "se" de la función geom_smooth
plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud") + theme(axis.title = element_text(face="bold"), axis.text = element_text(colour="black")) + geom_smooth(method=lm, se=FALSE)

#Para ajustar el color, podemos emplear el argumento "color" de la misma función
plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud") + theme(axis.title = element_text(face="bold"), axis.text = element_text(colour="black")) + geom_smooth(method=lm, se=FALSE, color="black")
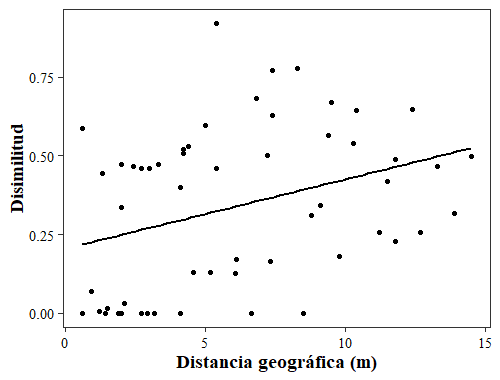
#El ajuste del grosor de la línea de tendencia se ajusta con el argumento "size" de la misma función
plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud") + theme(axis.title = element_text(face="bold"), axis.text = element_text(colour="black")) + geom_smooth(method=lm, se=FALSE, color="black", size=.5)

plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud") + theme(axis.title = element_text(face="bold"), axis.text = element_text(colour="black")) + geom_smooth(method=lm, se=FALSE, color="black", size=.5) + ylim(0,1)

plot <- ggplot(df, aes(x=Distancia, y=Disimilitud)) + geom_point() + theme_test(base_size = 14, base_family = "Times New Roman") + labs(x="Distancia geográfica (m)", y="Disimilitud") + theme(axis.title = element_text(face="bold"), axis.text = element_text(colour="black")) + geom_smooth(method=lm, se=FALSE, color="black") + ylim(0,1) + annotate("text", x=13.5, y=1, label="italic(r)==0.3423", family = "Times New Roman", parse=T, size=5)

¡Listo! Estos serían los ajustes principales que se le pueden hacer al gráfico. Las dimensiones que utilicé para exportar fueron de 400 (Width) x 380 (Height).
El plus:
¿Cómo podemos interpretar todo este revoltijo?
Sencillo. El valor r es el coeficiente de correlación. Recordemos que este valor oscila entre -1 y 1. Este valor sólo nos indica la intensidad y orientación de la correlación (positiva o negativa) que estamos probando. Si el valor es negativo, nos está indicando que la correlación es negativa, es decir, es inversamente proporcional. En el sentido contrario, cuando el valor es positivo, la correlación es directamente proporcional. Entonces los escenarios posibles son:
A medida que aumenta la distancia geográfica aumenta la similitud de la composición de especies entre las boñigas (esto sería si la correlación fuera negativa). Sin embargo, nuestro coeficiente de correlación fue positivo (0.34323), y el nivel de significancia fue por debajo de 0.05 (0.031968). Esto nos permite concluir que, cuando aumenta la distancia geográfica entre las boñigas, la composición de especies entre estas es diferente (son menos similares).
Chan chan... hermos terminado. Espero les haya sido de utilidad.
